Болезнь Фабри: от молекулярных основ к персонализированной терапии. История, причины, клиника, современная диагностика и лечение болезни Фабри
Раздел: Статьи
/
Общая врачебная практика.
/
Наследственные заболевания.
/
Болезнь Фабри: от молекулярных основ к персонализированной терапии. История, причины, клиника, современная диагностика и лечение болезни Фабри
Содержание
1. Введение: исторический контекст и эпидемиология болезни Фабри
2. Молекулярные механизмы и патогенез болезни Фабри
3. Клиническая картина болезни Фабри: возрастной полиморфизм
4. Диагностические стратегии
5. Современные подходы к терапии болезни Фабри
6. Мультидисциплинарное ведение
7. Заключение и перспективы
Болезнь Фабри (ангиокератома диффузная) представляет собой редкое X-сцепленное лизосомное заболевание накопления, обусловленное дефицитом фермента α-галактозидазы А. Впервые описанная независимо Иоганном Фабри и Уильямом Андерсоном в 1898 году, эта патология характеризуется прогрессирующим мультисистемным поражением с накоплением глоботриаозилцерамида в эндотелии сосудов, кардиомиоцитах, нейронах и почечных структурах. В настоящем обзоре детально анализируются молекулярно-генетические механизмы заболевания, особенности клинического полиморфизма у гемизиготных мужчин и гетерозиготных женщин, современные диагностические алгоритмы, включая методы ферментного анализа, биомаркеров и гистологической верификации. Особое внимание уделено инновационным подходам к терапии: фермент-заместительному лечению, фармакологическим шаперонам и перспективам генной коррекции. Приводятся данные последних клинических исследований и обсуждаются стратегии мультидисциплинарного ведения пациентов.
1.1 Историческая справка
Первые клинические описания болезни Фабри (БФ) появились в конце XIX века:
- 1898 г. — Иоганн Фабри (Германия) описал случай 13-летнего мальчика с ангиокератомами
- Одновременно Уильям Андерсон (Великобритания) представил аналогичное наблюдение
- 1963 г. — идентифицирован биохимический дефект (накопление церамидтригексозида)
- 1989 г. — клонирован ген GLA (Xq22.1)
1.2 Эпидемиологические особенности
- Распространенность: 1:40 000 — 1:117 000 живорожденных
- Гендерные различия:
- У мужчин: классическая форма с ранней манифестацией
- У женщин: вариабельный фенотип из-за случайного инактивирования Х-хромосомы
- Этнические особенности: описаны "горячие точки" в Италии, Португалии, Японии
2.1 Генетические основы
Ген GLA (7 экзонов) кодирует лизосомный фермент α-галактозидазу А. Известно >1000 мутаций:
- Миссенс-мутации (60%)
- Нонсенс-мутации (15%)
- Делеции/инсерции (20%)
- Сплайс-сайт мутации (5%)
Особый интерес представляют:
- Псевдодефицитные аллели (p.Asp313Tyr)
- Мутации с остаточной активностью (поздние формы)
2.2 Биохимический каскад
Дефицит фермента (<5% от нормы) приводит к:
1. Накоплению глоботриаозилцерамида (Gb3)
2. Вторичному отложению лизосомосвязанного глоботриаозилсфингозина (lyso-Gb3)
3. Активации патологических процессов:
- Эндотелиальная дисфункция
- Нарушение аутофагии
- Митохондриальный стресс
- Фиброз органов-мишеней
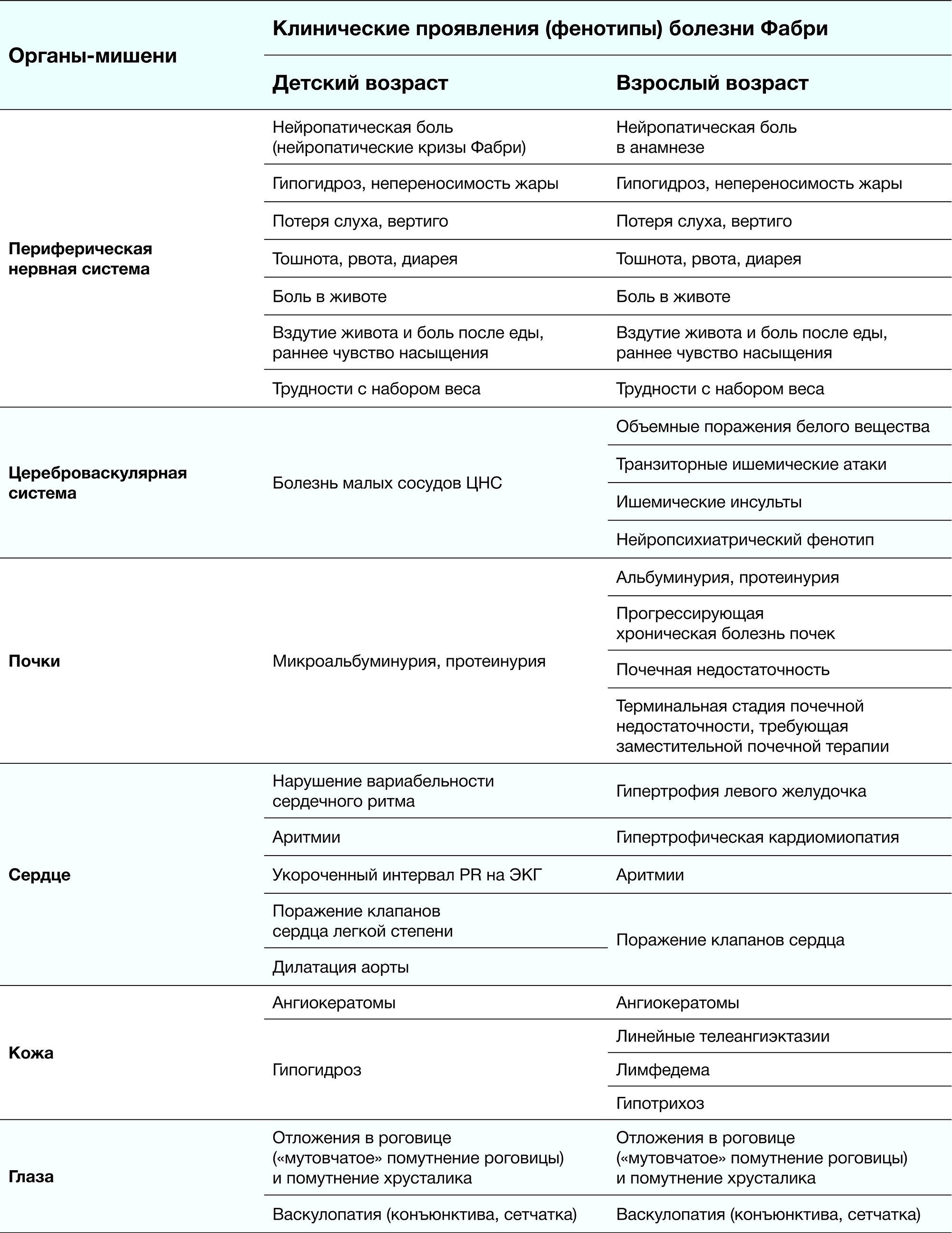
3.1 Классическая форма (мужчины)
Ранние симптомы (3-10 лет):
- Акропарестезии (жгучие боли в конечностях)
- Ангидроз/гипогидроз (95% случаев)
- Гастроинтестинальные проявления:
- Боли после еды (60%)
- Диарея/запоры (45%)
- Тошнота (30%)
Кожные проявления:
- Ангиокератомы (85%):
- Локализация: бедра, ягодицы, пупок
- Морфология: красновато-фиолетовые папулы
- Телеангиэктазии (60%)
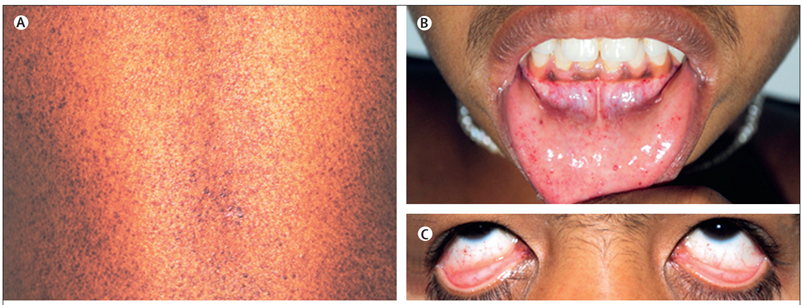
Поздние осложнения (20-40 лет):
- Почечная недостаточность (протеинурия → ХБП)
- Кардиальные нарушения:
- Гипертрофия ЛЖ (60%)
- Аритмии (20%)
- Цереброваскулярные события (15%)
3.2 Атипичные варианты
"Кардиальный" вариант:
- Дебют после 40 лет
- Изолированная гипертрофическая кардиомиопатия
- Отсутствие классических симптомов
"Почечный" вариант:
- Протеинурия как первый признак
- Быстрое прогрессирование до ТПН
Гетерозиготные женщины:
- Широкий спектр проявлений (от бессимптомных до тяжелых)
- Корреляция с паттерном инактивации Х-хромосомы
4.1 Лабораторная диагностика болезни Фабри
Ферментный анализ:
- У мужчин: активность α-Gal A в лейкоцитах (<3 нмоль/ч/мг)
- У женщин: недостаточно информативен (требуется генетическое подтверждение)
Биомаркеры:
- Плазменный lyso-Gb3 (чувствительность 93%, специфичность 98%)
- Повышенный Gb3 в моче
- Соотношение Gb3/креатинин
4.2 Генетическое тестирование
- Секвенирование нового поколения (NGS)
- MLPA-анализ для выявления делеций
- Пренатальная диагностика (CVS/амниоцентез)
4.3 Инструментальные методы
Кардиологическое обследование:
- ЭхоКГ: концентрическая гипертрофия ЛЖ
- МРТ сердца: позднее гадолиниевое усиление
- Холтер-ЭКГ: желудочковые аритмии
Почечная оценка:
- СКФ (цистатин С)
- Протеинурия (суточная)
- Биопсия: "мальтийские кресты" при поляризационной микроскопии
Неврологическое обследование:
- МРТ головного мозга: поражение белого вещества
- Исследование нервной проводимости (сенсорная нейропатия)
- Количественное тестирование потоотделения
4.4 Дифференциальная диагностика
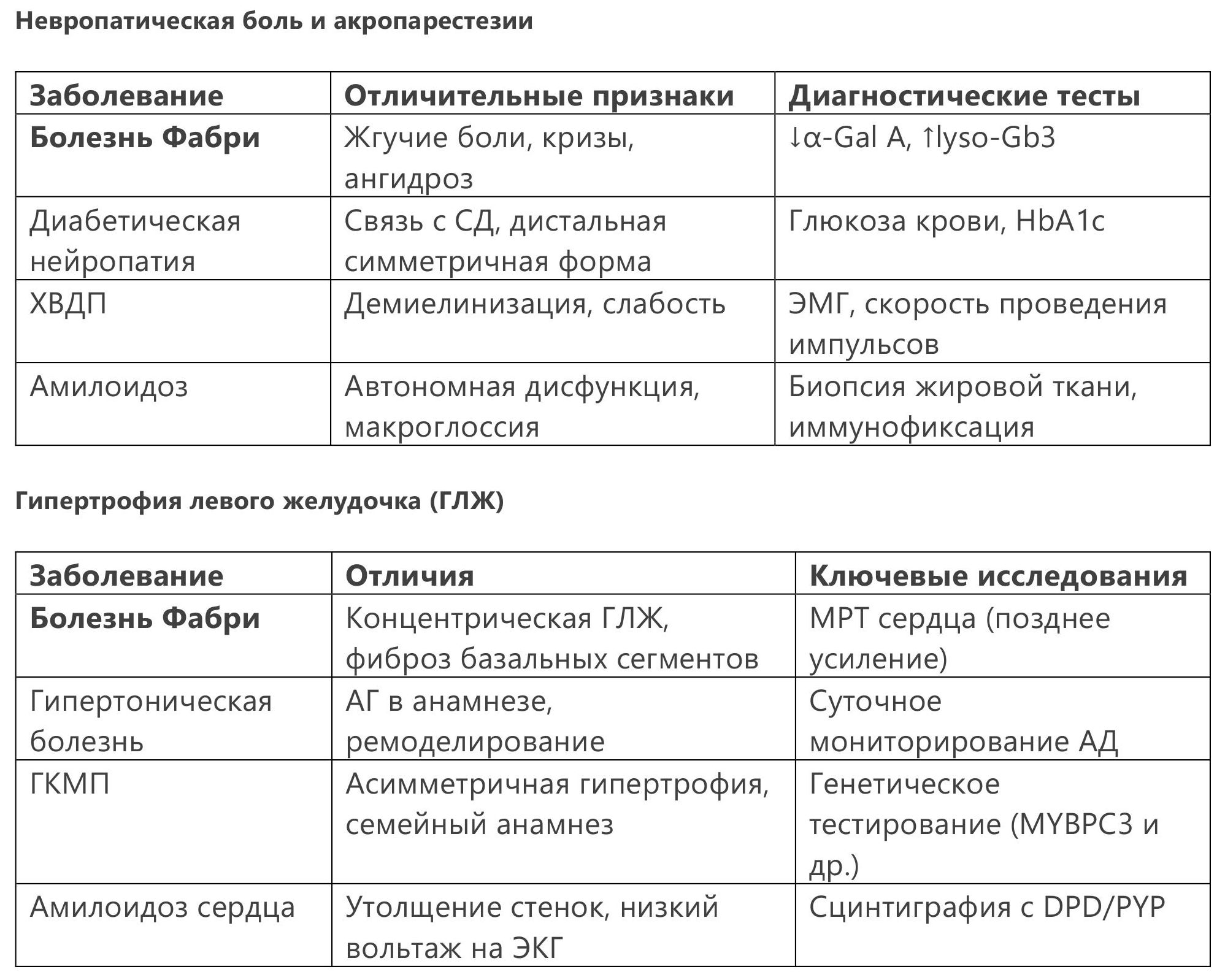
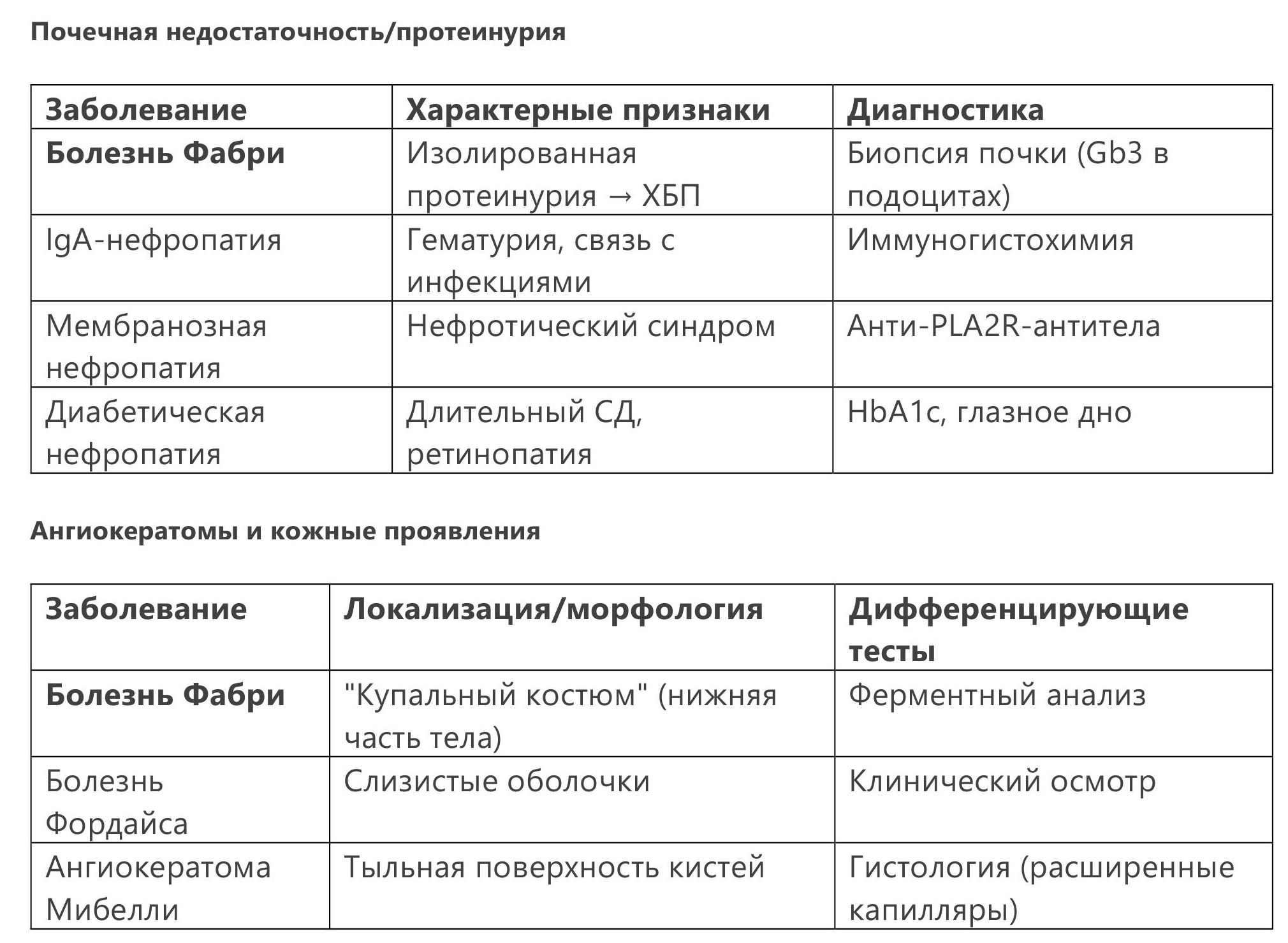
5.1 Фермент-заместительная терапия (ФЗТ)
Препараты:
1. Агалсидаза альфа (Replagal)
- Дозировка: 0,2 мг/кг каждые 2 недели
- Эффективность:
- Снижение lyso-Gb3 на 50%
- Стабилизация почечной функции
2. Агалсидаза бета (Fabrazyme)
- Дозировка: 1 мг/кг каждые 2 недели
- Особенности: более выраженный иммунный ответ
Проблемы ФЗТ:
- Формирование антител (40% пациентов)
- Ограниченное проникновение в некоторые ткани
- Высокая стоимость лечения
5.2 Фармакологические шапероны
Мигaластат (Galafold):
- Показан только для миссенс-мутаций (≈50% случаев)
- Механизм: стабилизация нативного фермента
- Дозировка: 123 мг через день
- Преимущества:
- Пероральный прием
- Лучшее проникновение в ЦНС
5.3 Перспективные методы
Генная терапия:
- AAV-векторы (исследования STARLIGHT)
- CRISPR-коррекция in vivo
Субстрат-редуцирующая терапия:
- Ингибиторы гликосфинголипидного синтеза
- Люциерастат (пероральный ингибитор)
6.1 Симптоматическое лечение
Нейропатическая боль:
- Прегабалин (75-300 мг/сут)
- Карбамазепин (200-400 мг/сут)
- Инфузии натрия тиосульфата
Кардиальная защита:
- Бета-блокаторы (метопролол, бисопролол)
- Ингибиторы АПФ (лизиноприл, периндоприл)
- Имплантируемые дефибрилляторы (при высоком риске)
Почечная заместительная терапия:
- Ранний гемодиализ
- Трансплантация почки (лучшие результаты с ФЗТ)
6.2 Мониторинг эффективности
- Ежеквартальное определение lyso-Gb3
- Годовое комплексное обследование:
- МРТ сердца
- Нейропсихологическое тестирование
- Оценка качества жизни (SF-36)
Несмотря на значительный прогресс в лечении болезни Фабри, остаются нерешенные проблемы:
- Преодоление гематоэнцефалического барьера
- Индивидуализация терапии на основе мутаций
- Оптимизация мониторинга отдаленных результатов
Перспективные направления включают:
- Развитие комбинированных подходов (ФЗТ + шапероны)
- Создание регенеративных технологий
- Улучшение скрининговых программ
13.04.2025 | 21:56:08